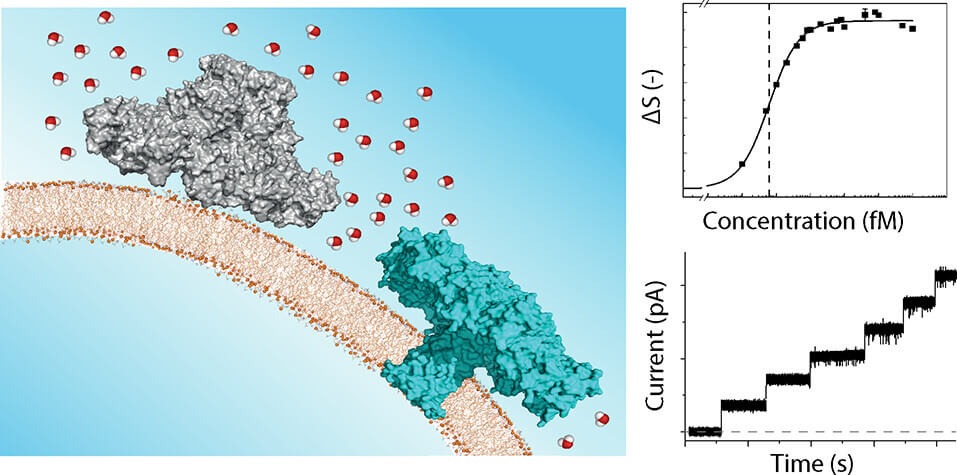
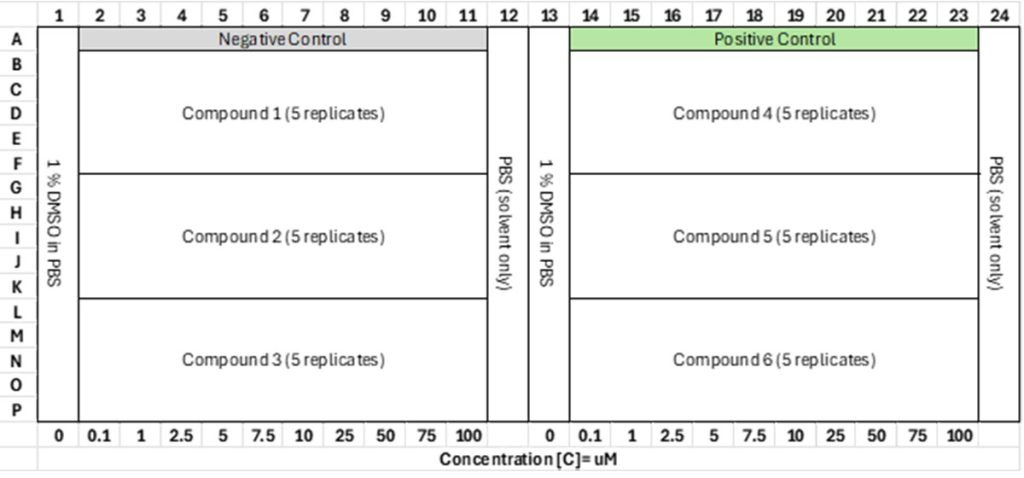
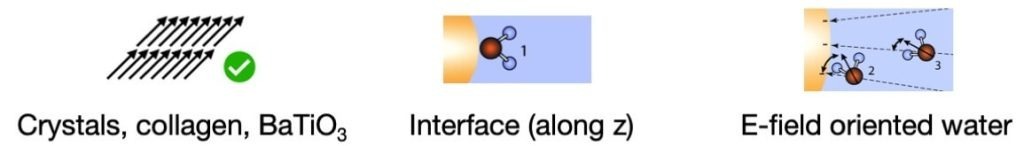
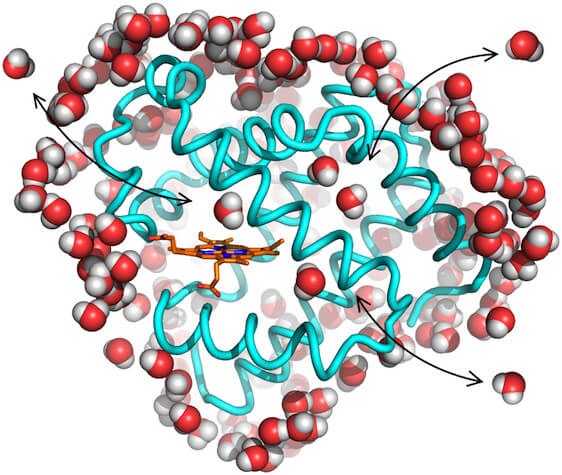
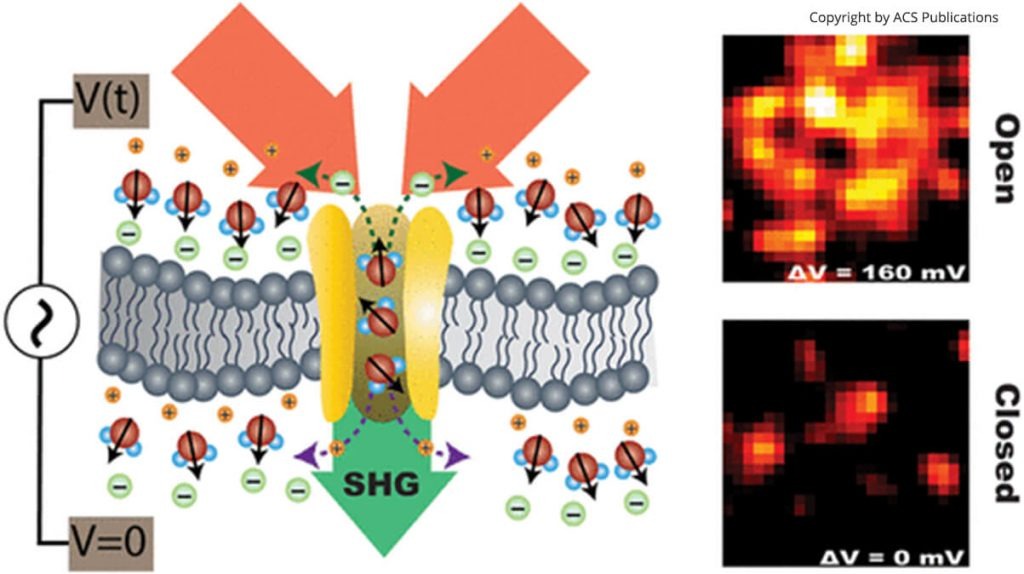
But as much as you are surrounded by air and you interact with it, you cannot say that it is not important to you. A solute molecule is interacting with the liquid around it, and this interaction goes both ways. The solvent is not only an inactive background. And this is especially true for water.
The solute is impacted by the solvent
The molecular interactions between the solute and the solvent have important consequences. Let us consider the most common solvent, water.
A very famous effect is based on the properties of water as a solvent: the so-called hydrophobic effect (water ‘repels’ substances that are similar to oil). It is key for phenomena such as phase separation or aggregation as it brings together hydrophobic entities, repelled by the water. This is one of the driving forces for holding together lipids that form cell membranes, one of the key brick of biological life on Earth.
This effect plays a role on the conformation of proteins in aqueous media. Depending on the environmental conditions, a protein will fold and unfold, hiding or revealing its hydrophobic cores and its binding sites, thus impacting its activity.
What is true for water, is also true for other solvents. In general, the solvent is responsible for ‘hydrating’ the solute molecule and it is a major factor in solubility measurement. This is why the solubility of a drug in pure water is different from the solubility in water mixed with DMSO, another solvent.
The solute molecule can also influence the liquid around it. But before that, we need to understand how the structure of the solvent works.
The structure of a liquid – how the solvent affects itself
A pure solvent is a liquid made of identical molecules, that move next to each other and are linked by a variety of forces. Different interactions (attractive or repulsive) are involved:
- the strong covalent/chemical bonding force (short range, attractive)
- the electrostatic interaction between charged molecules
- the charge-dipole interaction between a charged molecule and one that is uncharged but polarized
- the dipole-dipole interaction between two dipoles
- the hydrogen-bond, that is a strong type of directional dipole-dipole interactions
- the so-called Van-der-Walls forces that are weak and often called dispersion forces
- the repulsive forces (short-range), that prevents molecules to collide and fuse between each other
What you should remember here is that the molecules in the liquid will repel or attract each other depending on these interactions, in a very dynamic and fast manner.
These interactions, especially the short-range ones, give a very unique structure/molecular ordering to each liquid. For instance, strong hydrogen-bonding molecules can create dimers (e.g., fatty acids), linear 1D chains or rings (e.g., alcohols, hydrofluoric acid), 2D layered structures (e.g., formamide), or 3D structures (water).
And the liquid properties are determined by this ordering. For instance, if a liquid has a strong hydrogen-bonding, it will have a higher melting or boiling point (it will melt from the solid phase, or boil into a gas, at higher temperature). This is especially crucial in the case of water: its freezing/melting and boiling points make it perfect to be the ‘liquid of life’ on Earth.
The solvent is affected by the solute
The solvent structure rests on a subtle balance of the interactions mentioned above. But what happens when one introduces a solute (a different molecule) in it? The solute will add new interactions that will change the structure of the liquid and its properties.
A first example is when you add a charged solute to a solvent, such as salt to water: it does not freeze as easily as pure water. Sea water is very rarely frozen, will it is more common for the surface of a lake. This is because the addition of a salt that dissolves in water will introduce charged ions in the liquid. Then the charge-dipole interaction between the ion and the water dipoles will reorient the water molecules around it on a fairly long distance. This reorientation competes with the usual process of freezing (reorienting the water molecules to form an ice crystal) that happens usually at 0°C.
Confinement is another case of influence of the solute on the solvent. The insertion of a big molecule or an interface (oil droplet, lipid membrane, etc) that is insoluble in the liquid creates a boundary, a new frontier that blocks and alter the dynamics and structure of the liquid in its immediate vicinity. Confinement can be described as one-dimensional (next to a planar surface for instance), two-dimensional (water inside a pore or a nanotube), or three-dimensional (e.g., within a vesicle or a cell). Because of it, the solvent molecules experience less degrees of freedom to move, reorient, and interact with each other (it can make less hydrogen-bonds). Researchers have observed a broad range of effects caused by the solute such as slower relaxation times, changes in thermodynamics, freezing transitions, etc, compared to the bulk solvent far away from the solute.
Scientists found that these perturbations of the balance of interactions manifest on different time- and length-scales. They are stronger in the vicinity of the solute, and decreasing when going away from it, extending up to hundreds of nanometer in some cases.
However, investigating the solvent behavior at these scales is challenging, and this reflects in the plethoric lexical field used to interpret the influence of the solute. Here are some examples for the case of water:
- It is often stated that water gets an ‘icelike’ structure next to big solutes, or properties similar to cooled or supercooled water. While not completely wrong, this is misleading as these changed properties come not from the solid state of water (i.e. ice) but from the solute presence.
- In a similar spirit, some coined the term of ‘biological water’ for water molecules next to proteins. While water has indeed an extreme importance in biology, this tends to hide the fact that the solute molecule itself carries the biological function.
- Finally, the motion coupling of water and biological solutes has been described as water ‘slaving’ the solute motions. This is maybe a more accurate way of describing the mutual influences between the solute and the solvent molecules.
Studying these mutual influences between solvent and solutes is a challenge of extreme difficulty. Oryl has developed a groundbreaking technique to look at the reorientation if the solvent around a solute.
More information